NOTAS
Trabajos científicos
VER TODAS
MIOSITIS INFANTIL AGUDA BENIGNA
INFECCOIN URINARIA EN NIÑOS 2021
VITAMINA D Y EXPOSICIÓN SOLAR
VACUNA ANTIGRIPAL - TEMPORADA DE GRIPE EN EE. UU. 2019-2020:
AEROSOLES – GOTITAS (DROPLETS) - VIRUS Y TRANSMISIÓN
MIOPIA.
HERNIA UMBILICAL
ANQUILOGLOSIA O FRENILLO LINGUAL CORTO
SEMIOLOGIA RESPIRATORIA: RUIDOS PULMONARES
BRONQUIOLITIS AGUDA
BRUE: ACONTECIMIENTO QUE AMENAZA LA VIDA.
LECHES DE CONTINUACIÓN – NIÑOS DE 1 A 3 AÑOS
ALERGIA A ALIMENTOS
MICROBIOMA HUMANO Y SUS INTERACCIONES
DISLEXIA
DISPLASIA DE CADERA
TRAUMATISMO DE CRANEO LEVE.
CONSTIPACIÓN EN NIÑOS
¿QUÉ SE CONSIDERA FIEBRE EN PEDIATRIA?
INTRUDUCCIÓN DE ALIMENTOS COMPLEMENTARIOS – Y RIESGO DE ALERGIA
INFECCION DEL TRACTO URINARIO EN NIÑOS.
EL FRIO Y SUSCEPTIBILIDAD A LAS INFECCIONES RESPIRATORIAS
ENFERMEDADES AUTOINMUNES EN ENFERMOS CELIACOS
OBESIDAD INFANTIL
NOMOGRAMAS DE PERDIDA DE PESO TEMPRANO EN LOS RN ALIMENTADOS CON LECHE MATERNA
SINDROME DEL CROMOSOMA X FRAGIL
TOXICOS AMBIENTALES
RESPIRACIÓN BUCAL EN RN.
OBESIDAD EN NIÑOS
REFLUJO GASTROESOFAGICO
INFECCION URINARIA RECURRENTE: UN FACTOR DE RIESGO FRECUENTE EN NIÑOS
ENFERMEDAD CELIACA - PREVALENCIA Y CAMBIOS EN LA ALIMENTACIÓN
PROTECTORES SOLARES EN LA INFANCIA.
BRONQUIOLITIS - USO DE SOLUCION SALINA HIPERTONICA
VACUNAS ANTI- ROTAVIRUS Y RIESGO DE INVAGINACIÓN
ENFERMEDAD CELIACA: DIAGNOSTICO
LABORATORIO (3): INTERPRETACIÓN DE LAS PRUEBAS DE LABORATORIO
FIEBRE EN PEDIATRIA
¿PODEMOS DAÑAR LA PIEL DE NUESTROS NIÑOS?
ALIMENTACIÓN DURANTE LA DIARREA AGUDA
USO DE MACRÓLIDOS Y RIESGO DE ESTENOSIS HIPERTRÓFICA DE PÃLORO
IMPACTO DEL USO DE ATB, SOBRE LA RESISTENCIA BACTERIANA
LABORATORIO (2): ANALISIS DE ORINA EN NIÑOS.
LABORATORIO (1): INTERPRETACIÓN DE LAS PRUEBAS DE LABORATORIO MAS USADAS.
INFECCION URINARIA: RECURRENCIAS Y DAÑO RENAL.
ENFERMEDAD CELIACA - CLASIFICACIÓN Y NUEVAS PROPUESTAS.
NEUMONIA ADQUIRIDA EN LA COMUNIDAD:
USO DE BEBIDAS GASEOSAS PARA PREVENIR LA DESHIDRATACIÓN EN NIÑOS CON GASTROENTERITIS AGUDA.
VITAMINA D
ESPECTRO DE LAS ENFERMEDADES RELACIONADAS CON EL GLUTEN
ANOMALIAS RENALES PRENATALES/ MANEJO POST-NATAL
TRATAMIENTO DE FARINGITIS ESTREPTOCOCCICA EVALUACIÓN DE 3 TRATAMIENTOS DIFERENTES.
LA LECHE HUMANA (II)
LA LECHE HUMANA (I)
ALMACENAMIENTO Y CALENTAMIENTO DE ALIMENTOS LÃQUIDOS
CUANDO SE DEBE INTERNAR UN NIÑO CON ALTE?
EFECTOS DE LA MIEL EN LA TOS NOCTURNA.
INFECCIONES RESPIRATORIAS Y MASCOTAS
ALERGIA ALIMENTOS E IgE
INFECCION URINARIA EN LA INFANCIA: CONSECUENCIAS A LARGO PLAZO
FIEBRE Y ANTITERMICOS EN NIÑOS.
Diagnóstico visual
VER TODAS
“ECZEMA COXSACKIUMâ€
HALO DESPIGMENTADO ALREDEDOR DE UN NEVUS.
FACIES DE LLANTO ASIMETRICO
HEMANGIOMA SEGMENTARIO DE CARA
LACTANTE CON LESIONES NODULARES EN PIEL
PLAGIOCEFALIA DEFORMACIONAL.
HIMEN EN PEDIATRIA: POLIPO O TAG
LENGUA GEOGRÃFICA, PLICATA Y OTRAS.
PESTAÑA SUCIA
QUISTE DE RETENCIÓN MUCOSA: MUCOCELE
QUISTES PREPUCIALES DE QUERATINA
SÃNDROME DEL TORNIQUETE DE PELO
ALOPECIA TEMPORAL TRIANGULAR
Casos clínicos
VER TODAS
ADOLESCENTE CON DOLOR ABDOMINAL PERIODICO
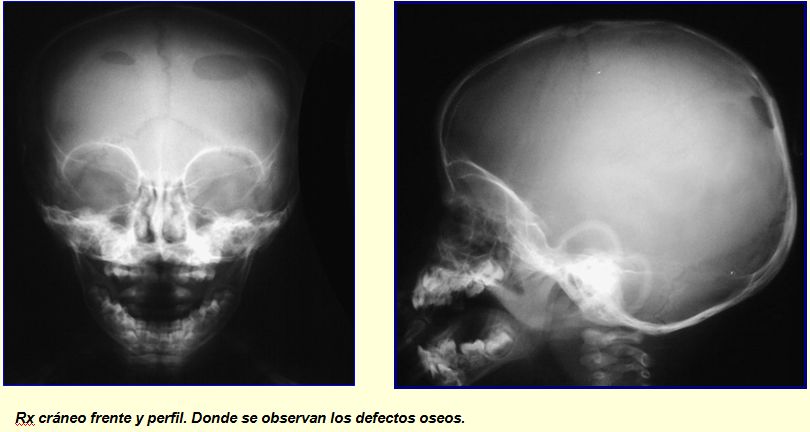
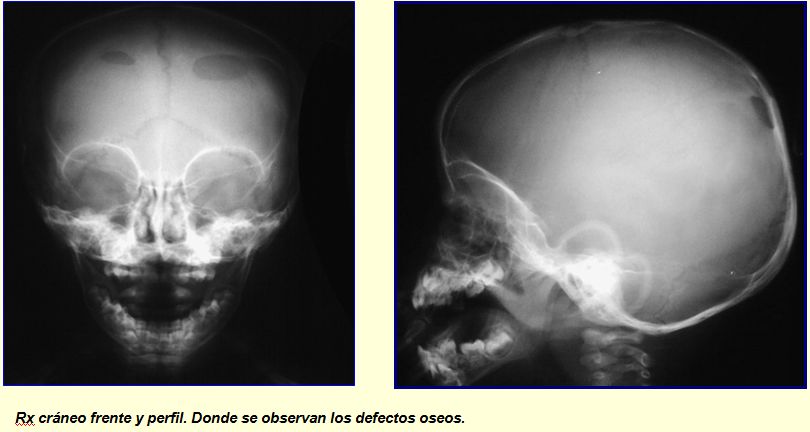
RECIEN NACIDO CON UN HUNDIMIENTO CRANEAL
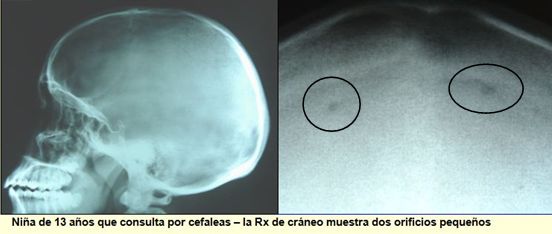
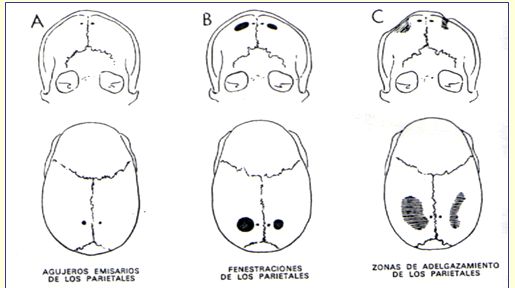
NIÑO CON DEPRESIONES EN SU CUERO CABELLUDO
NIÑA CON DOLOR TORÃCICO.
RECIEN NACIDO CON LESIONES REDONDAS EN PIEL.
UN NIÑO CON UN EXANTEMA EN TRONCO
NIÑO QUE SE NIEGA A CAMINAR.
UN NIÑO CON UN EXANTEMA MUY PARTICULAR
NÓDULO UMBILCAL PERSISTENTE EN UN NIÑO DE 6 MESES
LACTANTE DE 4 MESES CON BANDAS EN LA PIEL
PARCHE HIPERTRICOTICO - HIPERPIGMENTADO
LESION EN DEDO - HAGA SU DIAGNOSTICO
Alertas pediátricas
VER TODAS
ANDADORES
VACUNAS PARA LA GRIPE (2018)
CONTAMINACIÓN BACTERIANA DEL ESTETOSCOPIO
DIETA LIBRE DE GLUTEN: NO PARA TODOS LOS NIÑOS
LAS BACTERIAS DE LA PIEL PODRÃAN PROTEGER CONTRA ENFERMEDADES.
VACUNACIÓN ANTIGRIPAL
ENVOLVER AL BEBE, Y RIESGO DE SÃNDROME DE MUERTE SÚBITA INFANTIL.
ARSÉNICO EN EL ARROZ Y PRODUCTOS QUE CONTIENEN ARROZ
COLECHO / COHABITACIÓN
PARACETAMOL.
ANTIEMETICOS EN GASTROENTERITIS AGUDA EN NIÑOS.
CARACTERISTICAS SEROLOGICAS EN LA EFERMEDAD CELIACA EN NIÑOS –
EL SINDROME DEL BEBE “SACUDIDO†(SHAKEN BABY SYNDROME)
DISRUPTORES ENDOCRINOS AMBIENTALES. (DEA)
TRANSMISIÓN DE PERTUSIS MORTALIDAD ASOCIADA EN LACTANTES NO VACUNADOS.
DESAYUNO ABUNDANTE EN PROTEINAS EVITA EL AUMENTO DE LA GRASA CORPORAL EN ADOLESCENTES CON SOBREPESO.
RESPUESTA A LA VACUNA PARA HEPATITIS B EN CELIACOS
TE Y ABSORCIÓN DE HIERRO
RADIACIÓN POR ESTUDIOS DIAGNOSTICOS Y RIESGO DE CANCER.
USO DE TELEFONO CELULAR Y DISPOSITIVOS INALAMBRICOS – CONSECUENCIAS.
MELANOMA EN NIÑOS.
VACUNA GRIPE
CORDON UMBILICAL: ¿CUANDO LIGARLO?
PARACETAMOL DURANTE EL EMBARAZO
USO DE ANTIBIOTICOS EN LA INFANCIA Y RIESGO DE ASMA.
ESOFAGITIS EOSINÓFILA
VACUNA PERTUSSIS ACELULAR VS. CELULAS ENTERAS
LA ALIMENTACIÓN CON BIBERON AUMENTA EL RIESGO DE ESTENOSIS DE PÃLORO.
DIAGNOSTICO Y MANEJO DE LA OTITIS MEDIA AGUDA.
NUEVA VACUNA CONTRA MENINGOCOCO B
SÃNDROME DE APNEA OBSTRUCTIVA DEL SUEÑO. (SAOS)
USO DE ATB - β LACTAMICOS
METAHEMOGLOBINEMIA POR NITRITOS Y CONSUMO DE VEGETALES.
VACUNA GRIPE H1N1 Y NARCOLEPSIA
VACUNA ANTIGRIPAL: su efectividad
ACETAMINOFENO Y RESPUESTA INMUNE VACUNAL
RECOMENDACIONES DE LA FDA
NEUMONÃA E INHIBIDORES DE LA ACIDEZ GASTRICA.
Semiología
VER TODAS
PREPUCIO NORMAL Y PATOLÓGICO
ARRUGAS ACUAGÉNICA DE LAS PALMAS O QUERATODERMIA PALMO-PLANTAR ACUAGÉNICA,Y FIBROSIS QUISTICA
ADENOMEGALIAS EN NIÑOS
SINDROME DE TURNER.
NEVUS SEBACEO DE JADASSOHN
RN CON LESION EN PIEL Y TCS
DOLOR EN MIEMBROS INFERIORES.
DISRAFISMO ESPINAL OCULTO
TORTICOLIS EN NIÑOS
GENITALES FEMENINOS: FUSIÓN DE LABIOS MENORES:
FRENILLO LINGUAL CORTO O ANQUILOGLOSIA.
MARCHA EN PUNTA DE PIES IDIOPATICA.
MARCHA CON PIES HACIA ADENTRO
TORSION TIBIAL INTERNA (TTI)
MANCHAS MONGÓLICAS - SIGNIFICADO
CUIDADO DEL PREPUCIO
TESTICULO: CRIPTOQUIDICO CONGENITO - ADQUIRIDO – RETRACTIL.
OBSTRUCCION CONGENITA DEL CONDUCTO NASOLAGRIMAL
ALTERACIONES UNGUEALES EN NIÑOS
HEMANGIOMA INFANTIL Y HEMANGIOMA CONGENITO.
SIBILANTES MENORES DE 2 AÑOS Y USO DE BRONCODILATADORES
PIE PLANO FLEXIBLE
VELLO PÚBICO O ESCROTAL EN MENORES DE 1 AÑO.
PECTUS EXCAVATUM
SEMIOLOGIA DE LOS MIEMBROS INFERIORES
MAMELONES Y FOSITAS PREAURICLARES..
MASTURBACIÓN INFANTIL.
HIDROCEFALIA EXTERNA IDIOPATICA.
ACROPUSTULOSIS INFANTIL
Varios
VER TODAS
CESAREA VS. PARTO VAGINAL
SALBUTAMOL EN PEDIATRÃA
SINDROME DE PIERNAS INQUIETAS EN NIÑOS
PAROTIDITIS RECURRENTE JUVENIL
EL NIÑO QUE TARTAMUDEA.
INMUNODEFICIENCIAS PRIMARIAS ¿cuando sospecharlas?
TESTICULO NO DESCENDIDO – ACTUALIZACIÓN
CONDUCTA ANTISOCIAL Y TIEMPO DE TELEVISIÓN.
CHUPETE - Aliado o Enemigo
HIERRO: SUPLEMENTACIÓN Y TRATAMIENTO - EN NIÑOS
Información para padres
VER TODAS
PREVENCIÓN DE AHOGAMIENTO - BEBES NADADORES -CERCOS
TORTUGAS MASCOTAS Y SALMONELOSIS
HABLA Y LENGUAJE EN LOS NIÑOS
BABEO EN NIÑOS.
INGESTA DE SAL EN LA INFANCIA
CÓLICO DEL LACTANTE O LLANTO EXCESIVO
GASEOSAS Y ALTERACIONES DE LA CONDUCTA EN NIÑOS
PADRES QUE CHUPAN LOS CHUPETES DE SUS BEBES.
DISCIPLINA EN NIÑOS
INTRODUCCIÓN DE LOS ALIMENTOS COMPLEMENTARIOS
MEDIDAS PARA LOS PACIENTES CON INFECCIONES RESPIRATORIAS EN LA INFANCIA
NUEVA HERRAMIENTA PARA CRIBADO DE TRASTORNOS DEL ESPECTRO AUTISTA.
CUERPOS EXTRAÑOS NASALES EN NIÑOS
ASPIRACIÓN DE CUERPO EXTRAÑO EN NIÑOS.
COLECHO Y MUERTE SÚBITA
REPELENTES DE INSECTOS
IVERMECTINA PARA EL TRATAMIENTO DE LA PEDICULOSIS EN NIÑOS.
FIBRA DIETETICA - Información para padres
NECESIDADES DE HIERRO EN LOS BEBES Y NIÑOS. Información para padres.
DEPOSICIONES EN LOS LACTANTES – FRECUENCIA Y CARACTERISTICAS
VITAMINA D Y RAQUITISMO
"Saber no es suficiente, debemos aplicar. Desear no es suficiente, debemos hacer"
Goethe